

Reliance agreements have the potential to substantially reduce the administrative workload involved in multi-site research studies. Research shows that these agreements can lower overall costs and provide valuable support to institutions nationwide. However, implementation remains a challenge. A survey of 102 single IRB staff members from 20 different IRBs revealed that nearly all found the agreements to be complex, time-consuming, and difficult to develop.
We know how frustrating it can be to work through the reliance agreement IRB process. The silver lining is the regulatory backing from federal policies that support the use of a single IRB for multi-site studies. This rule lets the IRB chair or “designee” approve an IRB at the lead institution, which removes the need for extra IRB reviews. The NIH single IRB Policy now requires domestic sites that take part in multi-site studies with NIH funding to use one IRB for ethical review. In this piece, we’ll give you a detailed, step-by-step guide to make IRB agreements work in 2025. You’ll learn to tackle common challenges while staying compliant with current regulations. 45 CFR 46.114(b)
Your Step-by-Step Roadmap to IRB Agreement Success
- Understanding IRB Reliance Agreements in 2025
- Definition and Purpose of a Reliance Agreement
- Regulatory Basis: and NIH sIRB Policy 45 CFR 46.114
- When and Why Institutions Use Reliance Agreements
- Key Stakeholders and Their Responsibilities
- Role of the Reviewing IRB vs Relying IRB
- Principal Investigator Obligations
- Institutional Signatories and Legal Oversight
- Step-by-Step Process to Implement a Reliance Agreement
- Common Challenges in IRB Agreement Execution
- Best Practices for Efficient IRB Agreement Management
- Monitoring Compliance and Handling Amendments
- Conclusion
- FAQs
1. Understanding IRB Reliance Agreements in 2025
Reliance agreements serve as the lifeblood of modern research cooperation, especially when you have multiple institutions taking part in a single study. These agreements set clear protocols for IRB review that streamline research while you retain control of rigorous ethical standards.
2. Definition and Purpose of a Reliance Agreement
A reliance agreement works as a formal, written document that lets an institution involved in research delegate institutional review board (IRB) review to an independent IRB or another institution’s IRB. People know these documents by various names like IRB Authorization Agreement (IAA), Cooperative Agreement, or Memorandum of Understanding (MOU).
We created these agreements to eliminate duplicate IRB reviews when multiple IRBs have jurisdiction over the same multi-site research protocol. It also documents the authorities, responsibilities, and communication between the organization conducting the ethical review and the institutions relying on the reviewing IRB.
Reliance agreements can cover:
- Single studies
- Categories of studies
- All human subjects research conducted under an organization’s Federal-wide Assurance (FWA)
The relationship between participating entities often determines the type of agreement based on financial, legal, or shared work arrangements.
3. Regulatory Basis: and NIH sIRB Policy 45 CFR 46.114
Two main sources provide the regulatory foundation for IRB reliance agreements: 45 CFR 46.114 and the NIH Single IRB Policy.
45 CFR 46.114(b)(1) requires any U.S. institution engaged in cooperative research to rely upon approval by a single IRB for research conducted in the United States. This requirement became effective on, for research receiving original IRB approval after this date January 20, 2020.
The NIH Single IRB Policy took effect on January 25, 2018. It requires using a single IRB to review NIH-funded multi-site studies where each site conducts the same protocol with non-exempt human subject research. This applies to:
- Grant applications submitted after January 25, 2018
- R&D contract solicitations issued after January 25, 2018
- NIH intramural research studies with initial review after January 25, 2018
All but one of these regulatory requirements have exceptions:
- Cooperative research where law requires more than single IRB review (including tribal law)
- Research where a federal department or agency determines that a single IRB doesn’t fit the context
All the same, such exceptions rarely happen, and NIH doesn’t view cost as a compelling reason for an exception.
4. When and Why Institutions Use Reliance Agreements
Research teams from different organizations working together on human subjects’ research typically need reliance agreements. These agreements prove valuable for:
- Collaborative studies with investigators from two or more institutions
- Multi-site studies using identical research procedures at multiple locations
- Studies where different research activities happen at different sites
The drive to save resources and improve the IRB review process makes these agreements essential. Since January 20, 2020, most U.S. government-funded cooperative studies need single IRB review if they meet non-exempt human subjects research criteria and involve multiple sites.
Institutions think over several factors before entering a reliance agreement:
- The other IRB’s policies and procedures matching institutional standards
- Study risk level
- Funding source and prime awardee institution
- Human research activity’s location
- Staff involvement and expertise
- IRB expertise and most suitable IRB for the review
Effective implementation of reliance agreements helps institutions maintain high ethical standards while removing unnecessary administrative burdens in multi-site research.
5. Key Stakeholders and Their Responsibilities
The success of IRB agreements relies on clear role definitions for everyone involved in research collaboration. Good coordination between stakeholders helps maintain ethical research standards and reduces paperwork.
6. Role of the Reviewing IRB vs Relying IRB
The Reviewing IRB (also called the IRB of Record, single IRB, or lead IRB) has the power to review and oversee collaborative research projects. This IRB performs the regulatory review that other institutions will rely upon. The to report its findings and actions to officials at the relying institution and share relevant meeting minutes when asked Reviewing IRB follows written procedures.
The Relying IRB hands over review authority to the Reviewing IRB without re-examining the study. Both institutions aim to oversee ethical research conduct, but their practical effect is different by a lot. The Relying IRB must:
- Check if site investigators meet institutional requirements
- Make sure education, training, and qualifications are complete
- Share conflicts of interest and management plans with the Reviewing IRB
- Follow the Reviewing IRB’s decisions
An IRB can decide not to serve as the IRB of record for another location or rely on another IRB’s review.
7. Principal Investigator Obligations
The Principal Investigator (PI) has key responsibilities, whatever their institution’s role. The simple definition of a PI is “the project director or principal investigator and any other person, regardless of title or position, who is responsible for the design, conduct, and reporting of research”.
PIs at Reviewing Institutions need to:
- Submit all IRB-related materials on behalf of the relying sites, including initial applications, future continuing reviews, amendments, and reportable events.
- Make sure all relying on sites know their institution’s policies
- Keep communication open with Relying IRBs as the agreement requires
- Get approval for site-specific documents and share them with collaborating investigators
PIs at Relying Institutions should:
- Tell their local IRB about certain changes and updates even with external IRB oversight
- Follow the Reviewing IRB’s policies and requirements
- Complete all local ancillary committee approvals
- Submit protocols and consent forms to their home institution for administrative review
All PIs must get IRB approval before starting any nonexempt human subject research.
8. Institutional Signatories and Legal Oversight
Only authorized people can sign reliance agreements. Usually, the IRB Institutional Official or someone they choose can sign single IRB reliance agreements—researchers can’t sign these agreements for their institutions.
to the reliance process. The relying institution might face consequences if federal rules aren’t followed, even when another IRB handles the review. Most institutions want their legal team to review any reliance agreement before signing Legal review is vital. This review often looks at issues like personal injuries, insurance coverage, and liability.
Legal matters get complex because reliance agreements can determine which institution is responsible for:
- Injuries to research participants from negligent IRB review
- An institution’s financial and reputation costs
- Breaking federal requirements, which could stop federal funding
Legal teams spend lots of time and effort negotiating these agreements.
9. Step-by-Step Process to Implement a Reliance Agreement
Research institutions need methodical planning and execution to implement reliance agreements. A well-laid-out approach helps direct you through the complexities of multi-site research collaboration after you understand these agreements. This step-by-step guide shows you everything in establishing effective IRB agreements in 2025.
Step 1: Determine Involvement and IRB Oversight
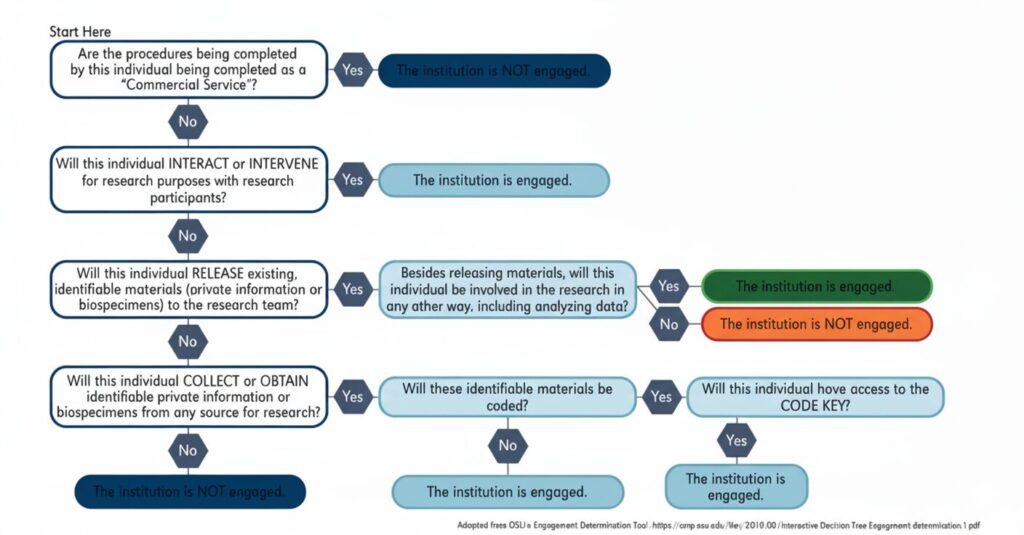
You should assess whether all participating institutions take part in human subject research. An institution takes part when its employees or agents: get data about subjects through intervention or interaction, access identifiable private information, or get informed consent from human subjects. This assessment is vital because it shows which institutions need IRB oversight.
Complex studies need you to think about:
- Which institution got the direct award from funding agencies
- Where subject interactions happen
- Who will access identifiable data
- Which site’s expertise fits the review
Your next step is to contact your institutional IRB office about a reliance arrangement’s feasibility before you approach potential collaborators.
Step 2: Drafting the Agreement Using SMART IRB Templates
The right agreement format comes after determining involvement. This has become the standard for new reliance arrangements since March 17, 2025 SMART IRB Reliance Agreement Version 3.0. This platform has several documentation options:
- Online Reliance System to coordinate multi-site studies
- Letter of Acknowledgement (LOA) for simpler arrangements
- SMART IRB Reliance Exchange (IREx) System for NIH-funded studies
SMART IRB agreements substantially reduce paperwork by eliminating individual agreements for each study. Institutions not using SMART IRB can negotiate alternative reliance formats during discussions.
Step 3: Legal Review and Institutional Approval
The agreement needs to be completed after the drafting phase. This vital step addresses liability concerns, including provisions for personal injuries and potential non-compliance with federal requirements institutional legal review.
Note that only authorized institutional officials, not investigators; can sign reliance agreements. The IRB Institutional Official or their authorized designee typically signs these arrangements.
Step 4: IRB Communication and Documentation
Clear communication channels between participating IRBs come next. The reliance agreement should spell out:
- The IRB responsible for initial and continuing reviews
- Procedures to submit amendments
- Protocol to report unanticipated problems
- Responsibility for conflict-of-interest management
- Local context information requirements
Each institution’s documentation requirements differ. Most reviewing IRBs need a completed local context form before they review your submission. You should register the study in your institution’s electronic IRB system even when using an external IRB.
Step 5: Finalizing and Archiving the Agreement
The final step happens when all parties approve the agreement and authorized officials sign it. The reviewing IRB sets the approval period and expiration date for the study.
Key documentation to archive includes:
- Fully executed reliance agreement
- IRB approval letters from the reviewing IRB
- Stamped consent documents
- Communication logs between institutions
These records should stay in a designated study-specific IRB binder throughout the study lifecycle for easy access during audits or inspections. Most institutions want you to after study completion keep these documents for at least five years.
10. Common Challenges in IRB Agreement Execution
Research teams face several major challenges when executing IRB agreements, despite careful planning. These obstacles can delay research and drive-up costs. Teams that understand these challenges can develop better strategies to address them.
Delays Due to Legal Review and Institutional Policies
Legal review stands as one of the biggest barriers to quick IRB agreement implementation. IRB professionals and members often point to “the lawyers” as a major roadblock to getting reliance agreements approved. The review process causes delays in multiple ways:
The legal review itself takes considerable time. Legal teams also raise concerns about specific provisions in reliance agreements, such as personal injuries and insurance coverage. This leads to lengthy negotiations. These negotiations happen because relying on institutions might face consequences if federal rules aren’t followed, even when another IRB handles the review.
These delays create substantial problems. A study revealed original reviews took 2 to 4 months. Full board reviews needed a median of 131 days, while expedited reviews took 85 days IRB review times. Multi-site studies face even greater complexity. A health services research study with 17 sites needed 115 submissions and used over 6,700 staff hours. The project ran almost two years longer than planned.
Variability in Agreement Language Across Sites
IRBs in similar situations often create very different reliance agreements. To cite an instance, two sIRBs supporting multi-site neurology trials funded by NINDS showed major variations despite their similar institutional contexts NeuroNEXT and Strokenet.
Key differences appear in these areas:
- NeuroNEXT requires relying on sites to handle HIPAA compliance, while Strokenet offers a flexible model for HIPAA issues
- NeuroNEXT makes the sIRB review permanent, but Strokenet doesn’t
- Both consider conflict of interest the relying site’s job, though NeuroNEXT can add more mitigation requirements
This variability creates problems because institutions usually want legal review of any reliance agreement they sign. The reviewing IRB prefers standard procedures to reduce administrative work, but the relying IRB wants to protect its interests through contracts. So, each new protocol often forces institutions to learn the process again.
HIPAA and Data Sharing Responsibilities
HIPAA compliance and data sharing create unique challenges in IRB agreements. The Privacy Rule requires covered entities to get IRB or Privacy Board approval to use protected health information without research participants’ authorization.
Several factors make this challenging:
The IRB or Privacy Board must document that any authorization waiver meets specific criteria. These include minimal risk to privacy, proper plans to protect identifiers, and written promises against misuse or disclosure.
Institutions must also decide how to share study-specific information between sites. This covers how to store enrollment lists and consent forms, and what happens if confidentiality gets breached.
Laws like HIPAA, FERPA, and GDPR often require contracts to share individual-level human subjects research data. Even with existing data-sharing contracts, researchers might need to send specific notifications or transfer data in certain ways.
The timeline to execute data sharing agreements depends on the institutions involved, data classification, and required consultations with other offices. Smart researchers develop their sharing plans during protocol development to avoid future delays.
11. Best Practices for Efficient IRB Agreement Management
Research teams need standardized tools and systems to manage IRB agreements well. These tools reduce administrative work and help teams work through complex multi-site projects more efficiently.
Using Centralized Platforms like SMART IRB
SMART IRB (Streamlined, Multisite, Accelerated Resources for Trials) platform stands out as the best way to coordinate reliance arrangements. The platform launched in July 2016 and makes it easier to adopt master IRB reliance agreements. It also provides resources that optimize documentation.
The SMART IRB Agreement works like a treaty between participating institutions rather than with one entity. Institutions can join by signing a “Joinder Agreement.” This eliminates the need to review, negotiate, and sign separate IRB reliance agreements for each study. The master agreement lets institutions rely on each other study by study and clearly defines everyone’s roles.
SMART IRB gives you several ways to document arrangements:
- The Online Reliance System (ORS) tracks requests and documentation
- Template Letter of Acknowledgement works for simple collaborations
- IRB Reliance Exchange (IREx) handles NIH-funded studies
Maintaining a Study-Specific IRB Binder
Regulatory binders store all required documentation for IRB reliance arrangements. These binders should have:
- Study protocol and synopsis
- Ethics committee approvals and supporting documents
- All correspondence between investigators and IRBs
- Sponsor correspondence or contracts
Electronic storage options have become popular. Teams can store documents in institution-specific study drives . Electronic documents can be shared with monitors through secure platforms like OneDrive.
The way you organize these binders matters a lot. A clear tabular system and keeping common sections at the front helps maintain order. Complex studies might need multiple binders with careful labeling.
Standard Operating Procedures for Multi-site Studies
SMART IRB Standard Operating Procedures (SOPs) guide reliance arrangements unless stated otherwise. These SOPs explain unique features of the SMART IRB Agreement and show how to set up reliant review of multi-site human research.
The Overall Principal Investigator must pick a Lead Study Team to work with the Reviewing IRB. This team collaborates to assign specific roles for sharing key information between institutions.
Of course, flexibility matters. The SMART IRB Agreement lets institutions decide who serves as the Privacy Board for HIPAA cases. They can choose to use combined or separate HIPAA authorizations and pick which entity reports to federal agencies. Teams can document these choices using SMART IRB’s implementation checklist.
12. Monitoring Compliance and Handling Amendments
IRB reliance agreements need continuous monitoring after they are put in place. Regular oversight helps teams stay compliant throughout the study and adapt to changes that come up.
Tracking Protocol Deviations Across Sites
Research sites must document and report protocol deviations promptly. These deviations fall into three main categories:
- Emergency deviations need reporting within 5 days to keep participants safe. Research teams must notify both the sponsor and reviewing IRB right away about these urgent changes.
- Major non-emergent deviations need IRB approval before teams can implement them. Any changes made without prior approval become non-compliance issues that teams must report quickly.
- Minor or administrative deviations usually only need to be included in summary forms at the time of continuing review.
Site monitoring often shows patterns that point to systemic problems. These patterns help teams figure out what protocol changes they need to make.
Notifying IRBs of Material Changes
Research teams must get IRB review for protocol amendments before making changes, unless they need to remove immediate risks to participants. Teams need to provide:
- A clear explanation of changes and why they’re needed
- Current enrollment numbers
- Plans to inform current participants
Even with external IRB oversight, investigators must tell their local IRB about certain changes, especially ones that affect ancillary reviews or data security.
Managing Informed Consent Across Institutions
Research teams must tell participants about important changes that might make them reconsider their participation. This includes:
- New risks or existing risks that have become more serious
- Procedure changes that affect participant’s time or effort
- Benefits that aren’t as good as predicted
Research teams should pick the best way to notify active participants based on how important the changes are.
13. Conclusion
IRB agreements change the map of multi-site research studies in powerful ways. In this piece, we looked at how these agreements make administrative processes smoother and managed to keep strict ethical standards for human subject research. Without doubt, 45 CFR 46.114 and the NIH Single IRB Policy give strong backing to these shared arrangements.
Research teams now have a clear path to handle reliance agreements. They need to figure out institutional engagement first to know which entities need IRB oversight. SMART IRB’s standard templates help cut down negotiation time. Legal review takes time but plays a key role in handling liability issues before making these agreements final.
Most institutions still don’t deal very well with these agreements. Legal reviews often slow things down. Different agreement language between sites makes it hard to standardize. It also gets tricky when dealing with HIPAA rules and data sharing – these need careful planning.
Smart practices help researchers get past these roadblocks. SMART IRB’s central platform cuts paperwork through master agreements. Good study binders, both paper and digital, help track everything during research. Special procedures for multi-site studies boost consistency and compliance.
Well-executed IRB agreements do more than just improve administration. Research teams can focus on scientific questions instead of paperwork, which helps them find answers faster. Study participants get the same protections at every research site.
Clinical research’s future depends more on smooth IRB processes to help multi-site teamwork. Challenges still exist, but the tools and frameworks in this piece give research administrators practical ways to set up good reliance agreements. These processes are worth learning for any institution that wants to work on shared human subject research.
To make your IRB management smoother and more efficient, explore our Fibi IRB Management System, designed to simplify the IRB management process.
14. FAQs
Q1. What are the key components of an IRB reliance agreement? An IRB reliance agreement typically includes the roles and responsibilities of the reviewing and relying on IRBs, communication protocols between institutions, procedures for submitting amendments and reporting issues, and provisions for managing conflicts of interest. It also outlines how HIPAA compliance and data sharing will be handled across sites.
Q2. How long does the IRB agreement process usually take? The timeline for implementing an IRB agreement can vary significantly, often taking several months. Legal reviews and negotiations between institutions are common sources of delay. For complex multi-site studies, the process may extend the overall study timeline by up to two years.
Q3. What is SMART IRB and how does it simplify the reliance process? SMART IRB is a centralized platform that offers a master IRB reliance agreement. Once an institution joins SMART IRB, it eliminates the need to negotiate individual agreements for each study. The platform provides standardized documentation options and procedures, significantly streamlining the reliance process for multi-site research.
Q4. Who is authorized to sign IRB reliance agreements? Only authorized institutional officials, typically the IRB Institutional Official or their designer, can sign reliance agreements. Investigators are not permitted to sign these agreements on behalf of their institutions. This ensures proper oversight and institutional commitment to the agreement terms.
Q5. How are protocol deviations handled in multi-site studies with reliance agreements? Protocol deviations in multi-site studies are categorized as emergency, major non-emergent, or minor/administrative. Emergency deviations require reporting within 5 days, major deviations need prior IRB approval, and minor deviations are typically reported at continuing review. The lead study team coordinates communication of deviations across all participating sites and with the reviewing IRB.


